Before we discuss cancer, let’s consider other sets of nasty characters – criminals, Nazis and terrorists. We know that the best way to limit crime is to create neighbourhoods that provide opportunities for education and jobs – a healthy infrastructure disfavours the criminal phenotype. Similarly, the Marshall Plan contributed to the rebuilding of Europe after WWII, likely preventing the emergence of another despotic regime (as the Nazis did in Germany after WWI, capitalizing on a country devastated after the war). The U.S.-led invasion of Iraq in 2002 had the stated goal of crushing terrorism, but instead left the country in such a devastated state that terrorism became a much bigger problem there than it was before the invasion. A devastated landscape is conducive to the terrorist phenotype. In each case, the numbers of potential criminals, Nazis, and terrorists born would have been similar, independent of the prevailing environment – the distribution of human genotypes is not the key variable here. Environmental disruption and instability favour phenotypes that we would consider as “maladaptive” in a healthy environment. In contrast, a stable healthy society is much more conducive to normality.
Early work from Beatriz Mintz, Mina Bissell and others has shown how the tissue microenvironment can substantially influence the cancer phenotype – the same cancer cells were shown to contribute to functional tissue phenotypes in a normal context, but form an invasive cancer in the wrong environment (1,2). In these cases, the environment directly alters the cellular phenotype. For the last 15 years or so, my lab has focused more on how environments either select for, or importantly, select against oncogenic phenotypes. If we want to understand why we get cancers, we should start by understanding why cancers are so rare during youth. We have argued that young, healthy tissues are inherently tumour suppressive, by disfavouring selection for oncogenic phenotypes (not so subtly presaged in the preceding paragraph).
You might ask – “how can this be? Oncogenic mutations will cause proliferation, even in a young healthy tissue. Don’t we avoid cancer in youth simply because insufficient mutations have accumulated?”While these views are widespread, they are wrong. I’ve dealt with the second belief extensively in previous critiques of this view (3,4), so we’ll focus on the first one here. That the occurrence of an oncogenic mutation should provide a cell-autonomous advantage is largely based on decades of research in “out of context” models, such as in cell cultures. Moreover, many mouse models engineer the oncogenic mutation in all of the cells of the targeted tissue (bypassing any need for the mutation to be advantageous). What happens when oncogenic mutations are created in normal stem cells in healthy tissues? For mouse hematopoietic stem cells (HSC), the result is quite reproducible across mutations disrupting tumour suppressor genes or activating oncogenes – these mutations cause loss of self-renewal (5). Loss of self-renewal is the kiss of death for a stem cell clone, leading to clonal exhaustion through differentiation (and the faster the rate of division, the faster the exhaustion).
We have argued that the same evolutionary force – stabilizing selection – that limits changes in organisms when environments are stable, also functions to limit somatic evolution in our tissues. In 1948, Herman J. Muller described how fruit flies maintain very constant features, such as the morphology and position of their wings (6). While it was very easy to identify fly mutants that changed these traits, in each case, mutational changes appeared to reduce fly fitness. He described the “high adaptive value of precisely the ‘normal’ degree of gene expression now existing” and that phenotypic stability in flies across many generations is “due to active selection in favour of the normal type”.
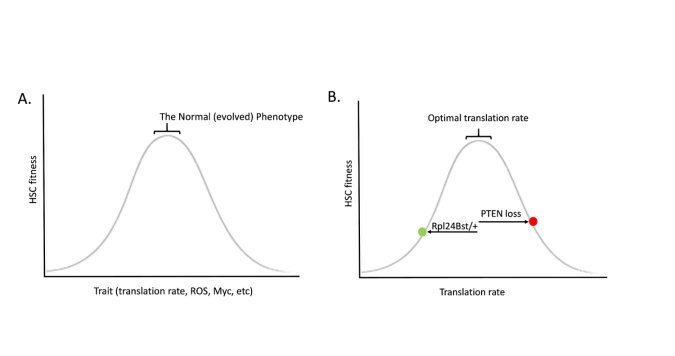
Just like a healthy forest will disfavour evolutionary change, so does a healthy tissue favour the status quo. Evolution is not progressive, and it does not seek any particular goal. Stabilizing selection is often depicted as a bell shaped curve, whereby decreasing or increasing a trait leads to reductions in fitness (Figure A). Perhaps the best demonstration for how such a relationship exists for HSC comes from the studies of Sean Morrison and colleagues on protein synthesis rates in HSC, showing that heterozygous mutations in a ribosomal protein (Rpl24Bst/+) reduce translation in HSC and impair HSC function and competitive potential (7). Morrison and others had previously shown that deletion of the PTEN tumour suppressor gene reduces HSC self-renewal (8,9); while the mutation was engineered in all HSC, were this mutation to occur spontaneously in a single HSC, it should almost always lead to clonal exhaustion (at least in a healthy HSC pool). PTEN loss also increases protein synthesis rates in HSC. Strikingly, combining PTEN loss with ribosomal protein mutation heterozygosity restores more normal translation in HSC, and also restores HSC function (7) (Figure B). Thus, both increases and decreases in protein synthesis rates impairs the ability of an HSC to be maintained in the compartment. Very similar relationships are evident for reactive oxygen species (ROS) (10) and Myc (11), where too much or too little of either leads to reduced HSC activity. Increasing translation, creating more ROS, and activating Myc – these are all common features of many oncogenic mutations, and they each have been shown to impair HSC self-renewal and/or competitive potential.
While some oncogenic mutations induced in HSC have been shown to increase competitiveness and self-renewal, I would argue that the methods used need to be considered. In particular, studies demonstrating that loss of DNMT3A and TET2 tumour suppressor genes increase HSC competitiveness have used high dose irradiation to condition recipient mice for these assays (12-18). I propose that these mutations will be either much less advantageous, or even disadvantageous, in healthy unperturbed bone marrow. Many other studies have utilized Mx-CRE to activate oncogenic mutations, which comes with the caveat that recombination requires induction of a potent interferon response (usually via injection of poly-IC). Ideally, we need to induce rare oncogenic mutations in healthy tissues without system disruption if we really want to understand how these mutations impact cell fate (not easy to do).
There are other ways to disfavour deviation from “normal”, including through induction of apoptosis or senescence following damage or aberrant activation of oncogenic signalling (19). Starting in the 1970’s, Gines Morata and colleagues described how cell competition in the fruit fly Drosophila melanogaster can eliminate cells with reduced function, such as through reduced protein synthesis or lower levels of Myc (20,21). Less competitive cells are actively eliminated (and even eaten) by their more fit neighbours. More recent studies have shown that too much of a normally good activity can be a bad thing too. Cells with oncogenic mutations in fly cells are actively eliminated by their wild-type neighbours (22,23), through well conserved signalling pathways. Again, the “normal type” is favoured. Similar mechanisms are evident in mammals, whereby an oncogenically mutated cell is forced out of a cell monolayer by its wild-type neighbours (24). For a cell in our airways or in our guts, such a fate would lead to expulsion of the covenant-disobeying cell into the outside world. A recent study further described how a normal tissue recognizes aberrant growths (whether due to impaired function or hyperfunction), with normal cells enveloping the mutant cells and expelling them from the tissue (25). Tumour suppression by eviction!
So basically, we are proposing that millions of years of evolution have selected for stem cells that are well-adapted to their tissue niches, which will strongly disfavour mutations that change phenotype. So why then do we ever get cancer? Importantly, the maintenance of healthy tissues, which favours the normal phenotype of resident stem cells, has been strongly selected by evolution through periods of likely reproductive success. However, as the odds of successful survival and reproduction (including the rearing of offspring for animals like humans) declines due to predation, low food availability, weather or other causes, the selective pressure to maintain tissues declines. So the same ideas proposed over 50 years ago to explain physiological decline in old age (26,27) can be applied to understand why cancer incidence also increases dramatically in older ages (28). The same oncogenic mutations that may be maladaptive in a healthy youthful tissue can become adaptive in an aged tissue, by providing an adaptation to the age-altered tissue landscape. Modern exposures, such as from cigarette smoke, pollution or alcohol, or more recent epidemics such as obesity, also clearly change tissue landscapes, and thus will radically change selective pressures. Hence, while it has been estimated that cancer incidence for most animals in the absence of recent disruption will typically be less than 5% (29), about 40% of humans will experience cancer, due both to longer lives and to modern exposures and lifestyles. In fact, just about everything we know that decreases cancer risk (exercise, a good diet, not smoking, etc.) is associated with healthier tissues – better favouring the normal phenotype for resident stem and progenitor cells.
While national security based on bombing other countries and “tough on crime policies” can earn you votes at home, building schools and infrastructure will produce better and less costly results in the long run. We similarly need to develop approaches to modify tissue landscapes such that the malignant phenotype is disfavoured, whether for prevention or treatment. Understanding that the fitness impact of a malignant phenotype is highly dependent on microenvironmental context should shift the emphasis towards targeting tissue landscapes, complementing therapies that are directly targeting malignant cells.
References
1. Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci USA 1975;72:3585-9
2. Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nature medicine 2011;17:320-9
3. DeGregori J. Connecting cancer to its causes requires incorporation of effects on tissue microenvironments. Cancer research 2017
4. Liggett LA, DeGregori J. Changing mutational and adaptive landscapes and the genesis of cancer. Biochimica et biophysica acta 2017
5. DeGregori J. Challenging the axiom: does the occurrence of oncogenic mutations truly limit cancer development with age? Oncogene 2012;32:1869-75
6. Muller HJ. Evidence of the precision of genetic adaptation. Harvey Lecture Series 1948;XLIII:165–229
7. Signer RAJ, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014;509:49-54
8. Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006;441:475-82
9. Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006;441:518-22
10. Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, et al. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxidants & redox signaling 2014;21:1605-19
11. Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes & development 2004;18:2747-63
12. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017
13. Kunimoto H, Fukuchi Y, Sakurai M, Takubo K, Okamoto S, Nakajima H. Tet2-mutated myeloid progenitors possess aberrant in vitro self-renewal capacity. Blood 2014;123:2897-9
14. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell 2011;20:11-24
15. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nature genetics 2012;44:23-31
16. Haladyna JN, Yamauchi T, Neff T, Bernt KM. Epigenetic modifiers in normal and malignant hematopoiesis. Epigenomics 2015;7:301-20
17. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proceedings of the National Academy of Sciences of the United States of America 2011;108:14566-71
18. Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer cell 2011;20:25-38
19. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004;432:307-15
20. Morata G, Ripoll P. Minutes: Mutants of Drosophila autonomously affecting cell division rate. Developmental biology 1975;42:211-21
21. Baker NE. Cell competition. Curr Biol 2011;21:R11-5
22. Ballesteros-Arias L, Saavedra V, Morata G. Cell competition may function either as tumour-suppressing or as tumour-stimulating factor in Drosophila. Oncogene 2014;33:4377-84
23. Menendez J, Perez-Garijo A, Calleja M, Morata G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proceedings of the National Academy of Sciences of the United States of America 2010;107:14651-6
24. Kajita M, Fujita Y. EDAC: Epithelial defence against cancer-cell competition between normal and transformed epithelial cells in mammals. Journal of biochemistry 2015;158:15-23
25. Brown S, Pineda CM, Xin T, Boucher J, Suozzi KC, Park S, et al. Correction of aberrant growth preserves tissue homeostasis. Nature 2017;548:334-7
26. Medawar P. An unsolved problem of biology. London: H.K.Lewis; 1952.
27. Williams GC. Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution 1957;11:398-411
28. Rozhok AI, DeGregori J. The evolution of lifespan and age-dependent cancer risk. Trends Cancer 2016;2:552-60
29. Hochberg ME, Noble RJ. A framework for how environment contributes to cancer risk. Ecology Letters 2017:20:117-134
comments powered by